
All you need to know about Gene Therapy
What is Gene Therapy?
Gene therapy is the delivery of a specific gene to correct or treat a disease.
The root of gene therapy can be traced back to the early 1970s when Stanfield Roger proposed that "good DNA" could be used to replace defective DNA in people with genetic disorders.
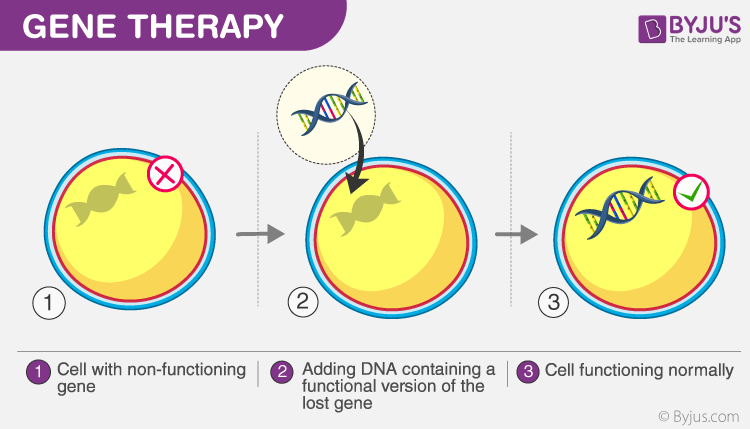
Gene therapies can work by several mechanisms, depending on the nature of the disease:
1) Delivery of functional genes into cells in place of missing/defective genes to correct a genetic disorder (Image above)
2) Inactivating a disease-causing gene that is not functioning properly or
3) Modifying a defective gene to treat or cure a disease
Gene Therapy Making the History
The first gene therapy was done in 1990 on a 4-year-old patient (Ashanthi De Silva). Ashanthi had adenosine deaminase (ADA) deficiency, a form of severe combined immunodeficiency disease (SCID).
Without ADA, T cells in the body die, leaving the individual unable to fight infections. Injections of a synthetic ADA enzyme help, but only temporarily. As such, a long-term solution is required. This is similar to most genetic diseases.

To treat Ashanti, doctors used a disabled virus as a carrier to deliver a functional gene to her cells. After successful delivery, Ashanti started to make ADA enzymes and lived a normal life.
On the back of this success, researchers started exploring the potential for gene therapy in other diseases. One of the main bottlenecks for gene therapy is the delivery of the new gene as it needs to enter the cell and make its way to the nucleus. To understand why it needs to go to the nucleus refer to our previous post on “Understanding DNA and the Central Dogma”.
Following the first success, doctors planned their second gene therapy treatment for Jesse Gelsinger. Gelsinger suffered from ornithine transcarbamylase deficiency, an X-linked genetic disease of the liver, the symptoms of which include an inability to metabolize ammonia – a byproduct of protein breakdown. However, subsequent gene therapy trials weren’t as successful. Like Ashanti’s case, doctors/researchers engineered a viral vector (A disabled virus carried the functional gene) to deliver the gene. But in Jesse’s case, the virus caused a systemic inflammatory response syndrome which led to his death.
Systemic inflammatory response syndrome is A serious condition in which there is inflammation throughout the whole body. Systemic inflammatory response syndrome may be caused by an infection, trauma, surgery, ischemia (lack of blood supply to a part of the body), or certain conditions, such as an autoimmune disorder or pancreatitis.
In Jesse’s case, the body started to fight the virus which was used to deliver the functional gene. Because doctors administered a very high dose of the disabled virus as the therapeutic the body mistook it to be an infection and mounted a strong immune response against it which led to disseminated intravascular coagulation and multiple organ failure causing his death. Following this event, the FDA put on hold all gene therapy programs until recently.
While the concept of Gene Therapy has been around for the past 30 years, the issue which has plagued gene therapy is finding a safe delivery method. This is one area that has seen tremendous research and success in the last 5 years.
Delivery Technologies
There are 2 delivery methods: viral and non-viral.
As the name suggests, viral delivery makes use of naturally found viruses in our environment which are exploited as carriers to deliver genes, similar to how a natural virus infects cells.
While viruses deliver their genes to different areas of the cell, for gene therapy the gene must get delivered to the nucleus. Several viruses allow for this, but a handful has been selected and are now the go-to viral delivery methods.
Similarly, non-viral delivery methods, as the name suggests are something other than exploiting a virus. On this front, scientists and researchers have developed synthetic nanoparticles which can be used for delivery.
One of the limitations though is that LNPs cannot deliver genes to the nucleus. As such, for gene therapies where a new gene needs to be introduced to replace a defective gene, this method would not work. However, LNP’s be used to deliver modalities to the cytoplasm which can then make their way to the nucleus to make corrections (think CRISPR/CAS9 or other editing technologies).
Viral Delivery
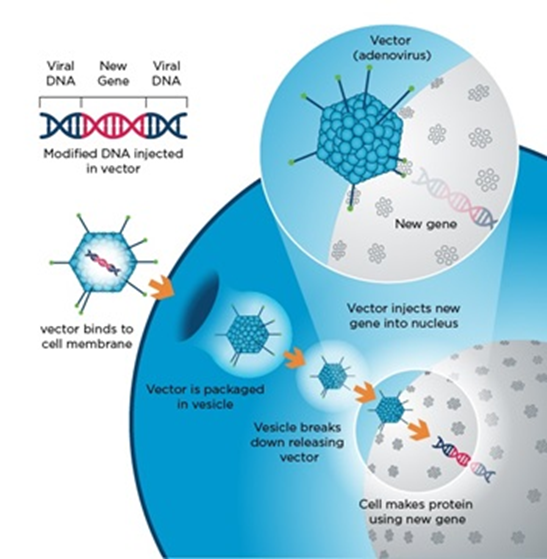
Viral vectors are tools commonly used by molecular biologists to deliver genetic material into cells. Competing for survival, viruses have developed capabilities to evade and enter host cells through different mechanisms.
The use of viral delivery methods is mimicking a virus infection. But instead of causing the host (that’s us) from falling sick, it delivers a modified functional gene and thereby replacing missing or defective genes. The image below provides explains the viral entry process into a cell.
Five of the most common viruses used for gene delivery are listed in the image below. These are viruses with which multiple labs, pharma, and other research organizations work. It is important to ensure that CDMOs have the capability or are willing to work with your virus as they will eventually need to be manufactured for distribution.
The below table was taken from Thermo Fisher Scientific (TMO) a well-known CDMO provider. You can read our deep dive on TMO here.

Using Ashanti’s case as an example, in this case, a retrovirus was used for delivery. The viral vector (disabled virus) is the 'car' used to transport the 'cargo' (ADA gene) into the patient's T cells ex-vivo (outside of the body).
After administration of the therapy, the viral carrier enters the cells and deposits the functional gene sequence into the nucleus of the target cells. This allows for the long-lasting expression of the ADA gene and the cells will continue to harbor a functional gene sequence. The genetically modified T cells then form part of the T memory repertoire (think of this like a computer backup, which can be accessed to copy the original file) which allows the body to continue to produce functional T cells, for up to 12 years.
Another example of a viral vector is the lentivirus. Lentiviruses are a subtype of retroviruses. The main difference between lentiviruses and retroviruses is that lentiviruses are capable of infecting both non-dividing and actively dividing cell types, while retroviruses can only infect actively dividing cells. In adults, most cells, such as myocytes, adipocytes, skin cells, and neurons, are non-dividing, i.e. terminally differentiated. As such the right type of virus must be picked depending on the target cells.
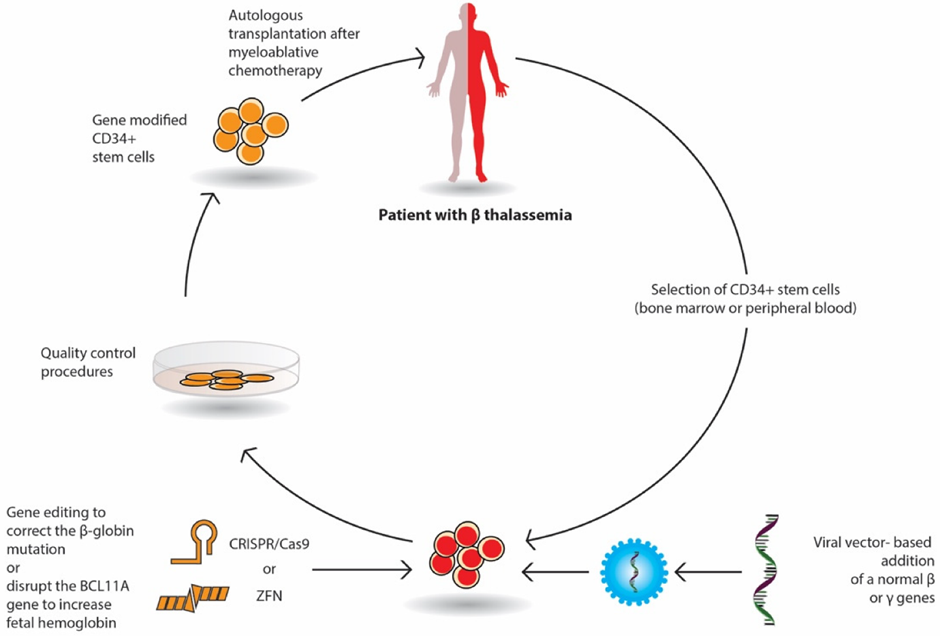
After almost two decades following Jesse Gelsinger's death, two of the lentiviral-delivered gene therapies were granted FDA approval. Bluebird bio’s (NASDAQ: BLUE) betibeglogene autotemcel (beti-cel) for severe β-thalassemia in August, followed by elivaldogene autotemcel (eli-cel) for cerebral adrenoleukodystrophy (CALD) in September 2022.
The image below shows how Bluebird bio’s beti-cel therapy is prepared.
One of the major drawbacks of both retroviral viruses and lentivirus is that they can integrate at random sites (although there is a 'hot spot' site where integration occurs more frequently). This means they can insert the genetic material which was delivered at a site that potentially disrupts a functional gene. This will lead to the loss of functional production of the protein (you just created a new problem for yourself) and can even cause cancer. The image below gives you a visual look at the integration process.
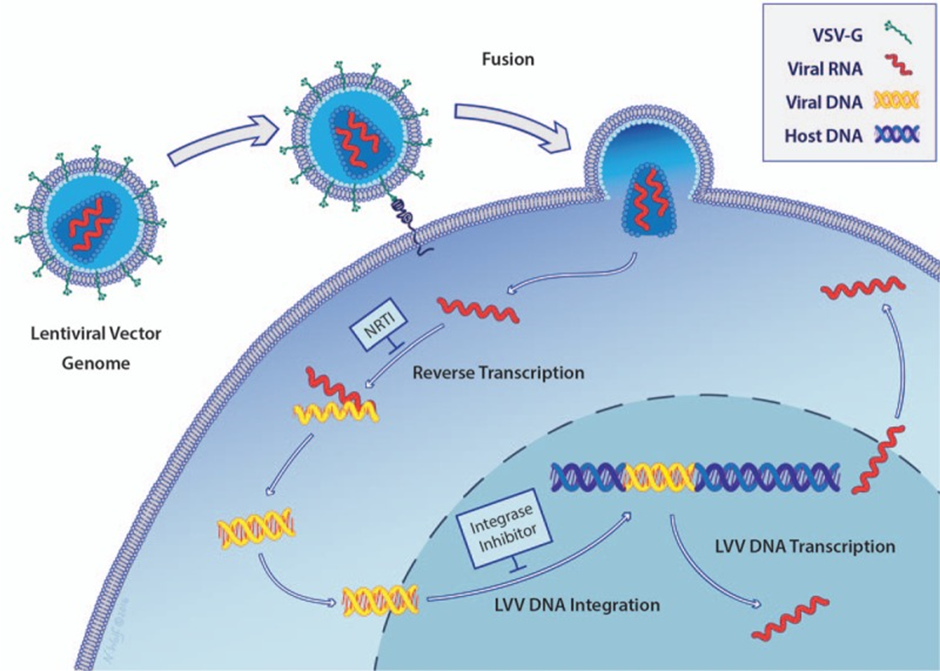
To bypass the potential for random integration, the researcher exploited and developed another delivery method using an adenovirus-associated virus (AAV).
Lab-engineered AAVs are non-integrating and additional manipulations to the virus shell ('capsid') can be made to improve the tissue-specificity ('tropic') of AAV. To be honest, I have chatted with friends working with AAV, they told me that although AAV is advertised to be non-integrating, there is a low but detectable level of integration. But as AAV integrates at a very specific site in the human genome, termed the 'AAVS1', the chance of disrupting a currently functional gene or cancer is rather low.
In 2017, Spark Therapeutics' Luxturna (RPE65 mutation-induced blindness) and Novartis' (NYSE: NVS) Kymriah (chimeric antigen receptor T cell therapy, CAR-T) were the first two AAV-based gene therapies to gain FDA approval.
Non-Viral Vectors
In the last decade, a variety of non-viral methods such as lipid nanoparticles (LNP) and extracellular vesicles have also been developed.
The mRNA SARS-Cov2 COVID vaccines developed by Pfizer (NASDAQ: PFE) and Moderna (NASDAQ: MRNA) are both encapsulated in LNPs. Without an LNP, mRNA will get degraded in our bloodstream before it can reach its target cell. The image below provides a graphical explanation.
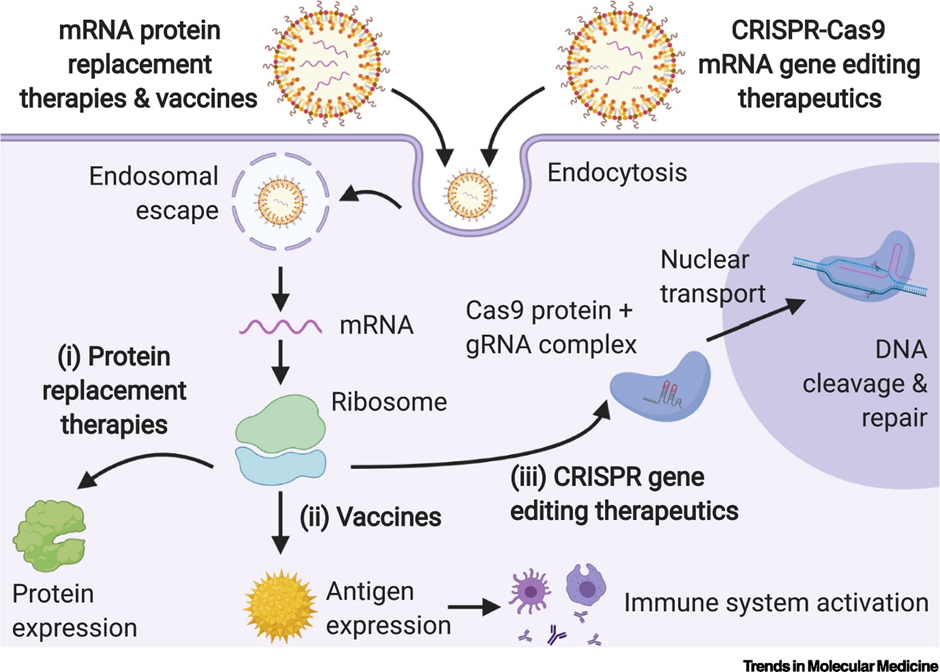
So how do LNPs work?
Cells are made up of bilayers of lipids which prevent molecules from entering cells. LNPs overcome this by allowing the fusion of lipid particles with the cell membrane. This allows the cargo (DNA/RNA) to enter into the patient cells, thus allowing the transcription and translation of therapeutic protein.
Apart from viral-based methods, LNPs are gaining prominence as a viable delivery method. Following its successful use to deliver millions of COVID-19 mRNA vaccines, this space has garnered a lot of research and commercial interest. It is estimated that the LNP CDMO market will grow at a CAGR of 13.8% between 2022-2030 as more mRNA, RNA, and DNA-based therapies enter the market.
Compared with viral vector delivery systems, non-viral delivery systems present a low risk for genome integration and cytotoxicity. However, it is less effective for targeted delivery and in delivering genetic material compared to viral vectors thus requiring a larger dose. This means more costs for manufacturing.
Another drawback of LNPs is that they tend to accumulate in the liver. That is why companies such as Alnylam (NASDAQ: ALNY) have been using their RNAi technology on liver diseases. You can read our deep dive on Alnylam here.
However, now companies are working on modifying surface charges, composition, and others of LNPs to allow for tissue-specificity. Particularly interesting are the modifications made to allow targeting of the lungs for cystic fibrosis by Moderna & Vertex Pharmaceuticals. You can read our deep dive on Vertex Pharmaceutical here.
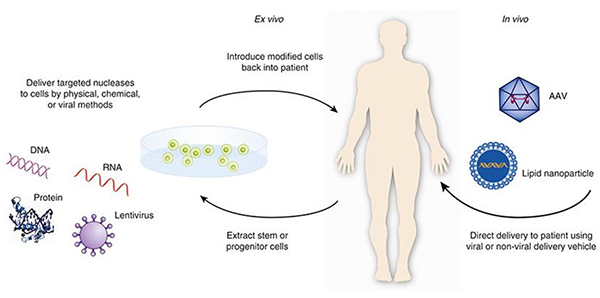
Ex-vivo vs In-vivo
Gene Therapy can be carried out via two routes. Ex-vivo or in-vivo. Let’s look at what that means.
Ex-vivo or "Outside the body" method is routinely used now. In this method, blood is drawn from a patient and the cell types of interest are isolated. These cells are then expanded and treated with the viral/non-viral vector. After this cells are purified selected for cells that have successfully been edited and to remove any excess virus/non-viral particles. Finally, the purified final product is injected back into the patient. One benefit of ex-vivo gene therapy is that it allows for greater control over which cells are injected back into the patient. This helps to reduce the potential risks associated with gene therapy. Some examples include sickle cell disease, adrenoleukodystrophy, chronic granulomatous disease, and others.
However, in some diseases, you cannot remove cells from the body to edit before putting them back. A good example will be some of the RNAi therapies which target the liver. Liver cells can't be removed and then reintroduced into the body. This challenge is also present for other organs such as the eye, lungs, etc. As such companies are testing an "inside the body" (in vivo) approach and will require direct IV infusion of the viral/non-viral based therapy into the bloodstream or injected directly into the target organ like the eye. For example, hemophilia and ornithine transcarbamylase deficiency (OTC) are good examples.
The below graphic shows the difference between the ex-vivo and in-vivo methods of therapy.
Current market landscape
There are a total of 27 approved Cell & Gene therapies at the moment. You can follow this link to see the whole list and read about each therapy if you are interested.
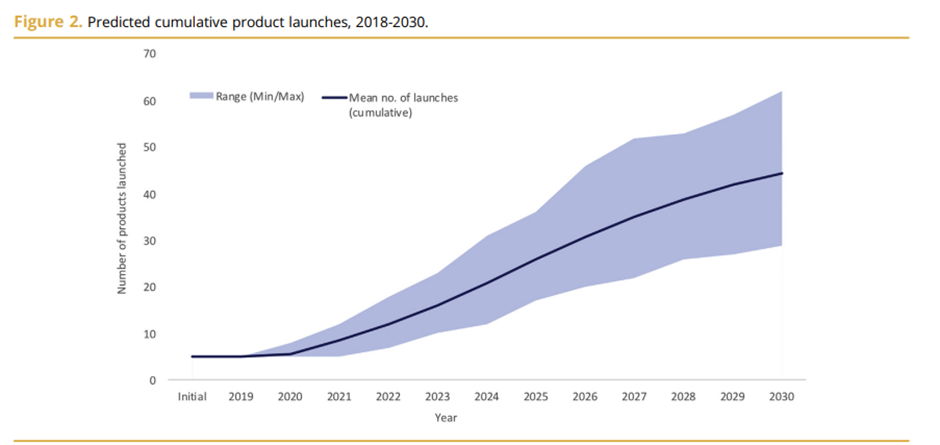
Over the next decade, it is estimated that we will see numerous C&G therapies entering the market. A paper in 2019 estimated that we could see close to 40 new products being launched per year in 2030. This will be across different indications with Hematological cancer leading the way with numerous therapies entering the market followed by Ophthalmology in a distant second place. The two graphs below provide details from the paper.
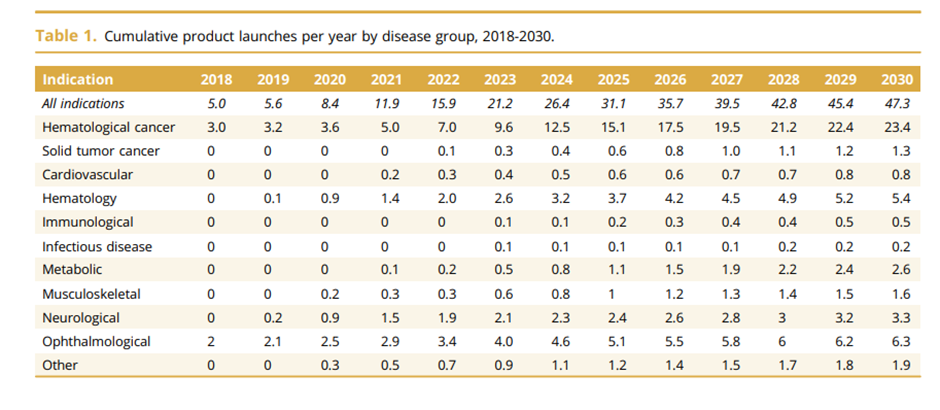
Looking more broadly, the C&G therapy market is estimated to be worth $10 billion by 2028 growing at a CAGR of 20.4%.
The next decade is going to be an exciting time for companies and investors as research performed over the last 30 years finally produces results in the form of approved therapies. As new therapies emerge it might lead to the growth of new biotech's and possibly new business models.
It's an exciting time and we are looking forward to seeing what the future holds.
If you are reading this, it probably means you enjoyed reading the post. We suggest you hit the subscribe button so you do not miss out on any of our future posts. Click here to Subscribe now.
Disclaimer: All opinions shared in this article are the opinions of the authors and do not constitute financial advice or recommendations to buy or sell. Please consult a financial advisor before you make any financial decisions.


Comments ()